
T-4- 103 – Conducting research under the Fertilizers Act
The 3 year regulatory transition period (October 26, 2020 to October 26, 2023) has now ended. As a result, regulated parties, including all manufacturers, importers, distributors and sellers of fertilizers and supplements must adhere to the amended Fertilizers Regulations. There are few notable exceptions for some product categories. Learn more about the implementation of the amended Fertilizers Regulations.
1. Purpose
The purpose of this document is to outline the requirements for fertilizers and supplements that are imported into or manufactured in Canada for experimental purposes. It also provides guidance on the process for obtaining research authorizations (RAs) for novel supplements under the Fertilizers Act and regulations.
2. Requirements for the import or environmental release of fertilizers and supplements in Canada
Most supplements and some fertilizers require registration and pre-market assessment prior to importation and sale in Canada. Companies sometimes opt to conduct research trials on fertilizers or supplements to evaluate their field performance in advance of submitting an application to register a given product under the Fertilizers Act. The Fertilizers Regulations contain provisions that allow persons to conduct scientific trials of unregistered fertilizers and supplements that would otherwise require registration prior to import or environmental release, as long as strict criteria are adhered to.
2.1 A person shall not manufacture, sell, import or export any fertilizer or supplement that contains any substance or mixture of substances in quantities that, if the fertilizer or supplement is used according to its directions for use, present a risk of harm to human, animal or plant health or the environment (except pests), if the fertilizer or supplement is used according to its directions for use, or in amounts not in excess of the amount that is necessary to achieve its intended purposes.
The objective of the regulatory requirements in place for research is to ensure that the environmental release of unregistered fertilizers (not exempt from registration) and novel supplements is safe, and to define conditions for product use, handling and disposal to reduce the risks to human health, animal health and the environment.
In some cases an import permit from the Animal and/or Plant Health Import/Export Office, Canadian Food Inspection Agency (CFIA), is also required. Please consult CFIA's Automated Import Reference System (AIRS) to determine if there are any further importation requirements.
Note: fertilizers and novel supplements that are imported under the research exemption provisions may not be used or distributed outside the confines of the research trial or offered for sale. Doing so is a contravention of the Fertilizers Act and regulations and may be subject to enforcement action.
3. Conducting research trials under the Fertilizers Act
3.1 Conducting research on fertilizers
A fertilizer that is imported into or manufactured in Canada for experimental purposes must be safe (section 2.1 of the regulations) but is exempt from registration and labelling requirements as long as:
- the product does not contain a supplement or a pesticide, and
- all treated plant material and all residual product are destroyed at the end of the trial so that they do not enter the commercial food or feed chains
Research trials conducted on fertilizers that are currently registered but for a different use pattern (for example ornamental plants vs. agricultural food crops) still require that all treated plant material be disposed of at the end of the trial to prevent their entry into the commercial food or feed chains.
Requirements for conducting research on fertilizers that contain supplements are the same as for novel supplements. Please see section 3.2 and 3.3.
3.2 Conducting research on supplements
Novel supplement means:
- a supplement that is not registered and not exempt from registration, or
- a supplement that is derived through biotechnology and has a novel trait
A novel trait, means a characteristic of the supplement that:
- has been intentionally selected, created or introduced into a distinct, stable population of the same species through a specific genetic change, and
- based on valid scientific rationale, is not substantially equivalent, in terms of its specific use and safety both for the environment and for human health, to any characteristic of a similar supplement that is in use as a supplement in Canada and is considered safe for use as a supplement in Canada
Research trials on supplements which are exempt from registration, or those that are already registered under the Fertilizers Act when conducted in accordance with the approved directions for use, is permitted without authorization from the CFIA. However, if the use pattern or directions for use of the product under test do not correspond to those approved, a research authorization is required prior to trial establishment. These types of authorizations are equivalent to testing novel supplements (see section 3.3 for details).
3.3 Research authorizations for novel supplements
Research authorizations are designed to ensure safety of the environmental release of the novel supplement; they prescribe confinement conditions, protective equipment, and crop disposal methods. The requirements and process for applying for a research authorization are detailed in the sections below.
4. Criteria for research trials
All research trials conducted on novel supplements in Canada must meet the following general criteria:
- trials must be conducted by a qualified researcher using scientifically valid methods. The researcher(s) responsible for the field trials must have successfully completed a Bachelor's or higher degree in agriculture or a related scientific field, or have previous experience in carrying out scientifically sound research trials on agricultural products
- good laboratory practices and appropriate quality control procedures must be followed. The company or research institute does not necessarily need to be certified for good laboratory practices
- the research institution must ensure the safety of its employees throughout the duration of the trial
- the research must be conducted in areas where spread of the product being tested outside the test site is not likely to occur (example: via run-off, erosion, drift, leaching, etc.) and bystander exposure to the supplement or to treated plants or soil is limited
- treated plants, growing media and unused product must not be sold or distributed and all supplement, fertilizer and plant material, including harvested crops, from the treated sites must be safely disposed of at the completion of testing, unless a waiver from the crop destruct requirement has been granted by the CFIA (please see below for details). Applicants are required to propose a method of safe disposal which may vary based on the nature of the supplement (microbial vs. chemical), the probability of its spread and establishment in the environment, and the relative risk associated with the release. For further guidance on safe crop disposal methods please contact the CFIA
5. Contained research exemptions and research waivers
Environment means the components of the Earth and includes:
- a) air, land and water
- b) all layers of the atmosphere
- c) all organic and inorganic matter and living organisms, and
- d) the interacting natural systems that include components referred to in a) to c)
Research trials where the release of the supplement into the environment is precluded, such as testing in contained facilities (laboratories for example), do not require a research authorization as long as appropriate measures are in place to prevent release into the environment. Research carried out in greenhouses may or may not be considered contained. To qualify as a contained trial, the conditions of greenhouse must be adequate to prevent the release of the novel supplement into the environment.
Applicants wishing to obtain an exemption from the research authorization for contained research trials must apply for a research waiver. The information requirements to obtain a research waiver include the following:
- name of the supplement or fertilizer. This includes a list of ingredients, their sources, concentration, guarantees and safety data sheets
- purpose and objective of the research
- proposed start date the research will take place
- the species of plants to be treated
- quantity of product required or imported for the experiment including rates, and method of application
- details outlining the disposal plan of the treated plants and product once testing is complete
- any other information relevant to the safety of the product. This may include a risk assessment and a description of personal protection equipment to be used while handling the product
- name, complete address and detailed description of the facility in which the product will be tested. This includes air handling systems, water and effluent handling systems etc.
- description of the treatment areas, isolation features and containment procedures
- handling practices to prevent the accidental release into the environment
- qualifications and training of the personnel
The confinement conditions of each facility are reviewed by the CFIA on a case by case basis and, if deemed appropriate, the exemption is granted. If there are any questions about how your facility would be classified, please contact the CFIA.
6. Crop destruct waiver
The crop destruct requirement may be waived by the CFIA if an appropriate comprehensive safety assessment (denoted as Data with Safety or DS) has been conducted, and the supplement or fertilizer has been deemed safe when used according to directions.
The safety assessment must be completed by the CFIA prior to submitting an application for a research authorization and a crop destruct waiver request. Applications for comprehensive safety assessment for the purpose of obtaining a crop destruct waiver will be placed in the file review queue and will not be accelerated. The CFIA's service delivery standard for a DS is equivalent to a new registration with data, found in Trade Memorandum T-4-122.
Applicants that have been granted a waiver from the crop destruct requirement may:
- conduct farm-scale trials (greater than 1 hectare)
- use farmer fields as opposed to research plots to conduct the testing; and
- use or sell the treated crop. Please note that crop destruct waivers do not authorize the sale and/or distribution of the supplement or fertilizer product itself
7. Overview of the research authorization process
7.1 Submitting an application
New research authorization applications can now be made through the electronic submission platform (MyCFIA). Applicants are strongly encouraged to use MyCFIA. While email or mail applications will still be accepted, they are slower to process and are therefore not the CFIA's preferred format/route from a file processing perspective.
The My CFIA digital service delivery platform allows you to request, pay for and track the status of services online anytime, anywhere through a secure account that can be tailored to suit your business model. To apply for a research authorization online, you must first create a My CFIA profile. Once enrolled, you will see the services available, including the Pre-market Application Submissions Office (PASO) service request of applying for a research authorization. Guidance documents, videos and step-by-step walk-throughs for how to sign up for an account, manage an account and request services online can be found at: My CFIA guidance.
If you choose to submit an application by email or mail, please submit the requested information to the PASO. When submitting by mail, it is preferable that the submission (or as much of it as is possible) be provided in electronic format (CD-ROM, DVD, flash-drive, etc.) to facilitate rapid processing of your application. The contact information for PASO is found in section 12.
Whether submissions are electronic or paper-based, the information required to be submitted in support an application is the same.
7.2 Application deadlines
All application packages must be submitted at least 90 days prior to the trial start date. This 90 days is the time required to process an application and does not include industry response time (for example the time during which a file is on hold waiting for payment, additional information, clarification or confirmation from the applicant). If submissions are received less than 90 days prior to the trial start date, the CFIA cannot guarantee that review of the information will be completed in time for the planting season.
7.3 Application review process
All applicants requesting a research authorization are required to fill out the application form (CFIA/ACIA 5475) providing information about the trial and the product to be tested. Based on the research category (description of each category is included in section 8), applicants are required to submit all the required information as detailed below. When received by the CFIA, the research authorization category will be confirmed and the applicant will be informed in writing of the decision and the appropriate fees (link to fees notice). This stage of the review process will be completed within 30 days of receipt of the application.
Once the fees are received and the information provided satisfies the requirements of the regulations, the next step of the review will commence. The CFIA will provide a draft appendix of the RA to the applicant to ensure its accuracy and to confirm that it reflects the proposed field trial plan. Any modifications or changes made by the applicant will then be assessed by the CFIA and the research authorization will subsequently be finalized.
Applicants will then receive a research authorization which describes the specific conditions of the trial. The conditions prescribed in the authorization must be adhered to throughout the duration of the trial. In the case of renewals, applicants will receive a research authorization describing the same conditions as those set out in the previous year (unless new information merits a change).
Any amendments to the final research authorization must be submitted to the CFIA for approval prior to implementation. This includes any changes to the research protocol such as the trial location, size, and /or methods of crop disposal etc.
Please note that where authorization is required, research trials cannot commence before receiving the research authorization and the trials must be conducted in accordance with the conditions prescribed therein. In all cases, the quantity of the product imported shall not exceed the total amount required to conduct the testing.
7.4 Required frequency of application for research authorization
For all categories of authorizations, an application form must be submitted for every year of the testing for each product and crop species or crop group.
Crop groupings:
classification of crops in agriculture and horticulture vary significantly depending on the criteria used (taxonomy, plant morphology, economical/commercial uses and agro-botanical characteristics).
Provided that the product, trial method and crop species or crop group are the same, trials conducted at multiple sites may be reported under the same research authorization. If applicants wish to report multiple trials conducted on crops from the same crop group under 1 application, a scientifically valid rationale demonstrating crop similarity must be submitted to and approved by the CFIA. This rationale should include taxonomic or otherwise physiologically relevant evidence indicating that the crop species within the group will respond to the novel supplement or a fertilizer containing a novel supplement under test in a similar manner. Please note that obtaining a research authorization for a crop group does not automatically guarantee that the supplement will be granted registration for that crop group. Safety data may be required for each crop species individually.
8. Categories of research authorizations
| Category | Description |
|---|---|
A: Standard research application for new research |
|
B: Research application for new research plus a safety review |
|
C: Renewal of a research authorization |
|
9. Documentation required, based on research authorization category
To apply for research authorization, all applicants must submit documentation to the CFIA according to the category of research authorization.
| Category | Documentation required |
|---|---|
A: Standard research application for new research |
Sections 9.1-9.8 inclusive, as applicable for a specific product |
B: Research application for new research plus a safety review |
Sections 9.1-9.16 inclusive, as applicable for the specific product |
C: Renewal of a research authorization |
Sections 9.1-9.3 inclusive |
9.1 Application form
All applicants are required to complete the application for research authorization form (CFIA/ACIA 5475). This form must be filled out in its entirety and signed by the applicant and the responsible researcher.
9.2 Experimental-use label
An experimental-use label or information sheet(s) is required to accompany the test product and must be included together with the application form. The experimental-use label or information sheet ensures that the applicators of the product are provided with the appropriate use, handling and disposal instructions. These labels or sheets must also be provided to the researcher in charge, and made available to all workers who will be directly involved in the trial. Once approved by the CFIA, the experimental-use label or information sheets must be affixed to the product container(s) to be used during the trial. The information should be legibly typed in both official languages and should correspond to the directions for use in the intended research project.
The following information is required to appear on the experimental-use label or information sheet accompanying the product.
Required statements:
- read label before using
- for experimental use only
- not for sale
- not for distribution to any person other than a researcher or cooperator
- any unused product should be returned to the manufacturer unless appropriate sterilization methods are available
Required information:
- name, product identifier
- name and address of the manufacturer
- net contents
- active ingredient and concentration or guarantee
- directions for use including crop species, method of application and application rate
- method of cleaning or sterilization
- crop disposal method (must correspond to the method prescribed in the research authorization)
- name and contact information for the researcher in charge and cooperator
Add to experimental-use label if applicable:
- storage requirements, expiry date
- product disposal
- hazard warnings and precautions
- first aid information
- personal protective equipment during transport, mixing, loading, application, disposal and spill cleanup
9.3 Trial maps
It is the responsibility of the applicant to provide the CFIA with the geographical location of the trial site including legal land descriptions and detailed maps. General test locations may be provided with the application forms, but the exact maps must be submitted no later than 21 days after trial establishment. Maps of all field trials must be legible and precise so as to allow CFIA inspectors to locate the trial site this year and in subsequent years, if need be. Maps should be on a blank page with crisp line drawings and block letters. Maps on lined or graph paper, or photocopies of road or topographical maps will not be accepted. The following information must be clearly printed on each map:
- the location of the trial site (city, town, province). For trial sites established within town/city limits, include the physical street address. For trial sites established outside town/city limits, include the distances and directions from the nearest town/city, as well as legal land description
- which direction is North indicated at the top of the page
- GPS co-ordinates of the trial site and 3 permanent surrounding landmarks. If this is not possible, a minimum of 3 separate measurements from 2 permanent nearby landmarks to 2 of the site corners (to the nearest ½ meter) must be provided for precise location of the site
GPS coordinates can also be used to mark the outer limits of the test area (example; for a rectangular plot, at the 4 corners) and 3 permanent surrounding landmarks. The permanent markers (landmarks) must be found within a 10km radius of the field site. If using a building or other large object as your permanent marker, please be specific as to which part of the building was used as a reference (example; south-west corner of the red barn). In addition, please choose well-spaced landmarks (do not take 3 reference measurements from the same building). Please note, that maps are still required even if trial locations are identified using GPS coordinates (the GPS coordinates should be marked on the map)
- exact trial dimensions and a description of surrounding crops
- the name and phone number of the field contact
- the Submission Control Number designated by the CFIA (required on the final map only) and treatments present at that trial location
- the date of the trial establishment
An example of an acceptable map (with and without GPS coordinates) is provided in Appendix A of this document.
9.4 Trial design
A description of the trial design and treatment regime is required for trials categorized as:
- Standard research application for new research, and
- Research application for new research plus a safety review
This includes the total trial size and the individual plot size(s), description of the treatments, number of replicates, seeding rate, and the specific details on the rates and method of application of the product. Buffer areas surrounding and between plots may also be required to mitigate the spread of the novel supplement or the fertilizer containing a novel supplement into the environment.
9.5 Constituent materials
All ingredients must be identified including the source and proportion of these materials. Safety data sheets (SDS) should also be provided for individual ingredients and final product.
9.6 Microbial inocula
For products that contain naturally occurring viable microorganism(s) the following information is required:
- purpose of the microbial strain
- taxonomic identification of the microorganism to the genus and species level; subspecies, and strain (if available)
- analytical results substantiating the taxonomic ID and classification
- methodology used to identify all microorganisms found in the final product
- relationship to known pathogens (phylogenetic trees)
- origin of the microorganism (when, where and from which material it was isolated). If the strain has been deposited or obtained from a recognized culture collection, for example American Type Culture Collection or other the bank accession numbers
9.7 Manufacturing process and quality control procedures
A brief description of the manufacturing process accompanied by a flow chart used as an overview of the process, together with the quality control procedures implemented to ensure purity of the final product and consistency in production is required. If the information is not available to the applicant, it may be provided directly by the manufacturer of the product. In such cases confidential business information shall not be disclosed to any person or party including the applicant without written authorization of the manufacturer.
9.8 Crop and product disposal
All plant material, including harvested crops from the treated sites, must be safely disposed of at the completion of testing, unless a Crop destruct waiver has been granted by the CFIA. Applicants are required to propose a method of safe disposal, along with a rationale based on the nature of the product (microbial vs. chemical), the probability of its spread and establishment in the environment, and the relative risk associated with the release.
Product disposal often involves returning all unused product to the manufacturer for proper destruction (autoclaving, incineration, etc.).
Crop destruct methods include, but are not limited to: collection and sterilization of all treated plant material, incineration, landfill disposal, harvesting and burning plant residual material on site, deep burial to a depth of greater than 1 meter, incorporation into the soil, disposal in accordance with standard provincial and municipal regulations and others.
9.9 Safety rationale
A safety rationale, based on experimental data and/or published scientific literature must be provided for the product and all its ingredients. In this rationale, please identify any potential risks the novel supplement or fertilizer containing a novel supplement may pose towards human health and the environment and discuss all studies where adverse effects associated with the use of the active ingredients were reported.
Please note that this safety rationale is not considered a "full safety assessment" for product registration and commercial release. Additional information may be required at the time of product registration, or for a crop destruct waiver for certain products
9.10 Microbial hazard characterization
Through publically available or primary data, please address the following hazard endpoints:
- human: pathogenicity, toxicity, sensitization/irritation and dermatophytic potential
- terrestrial plants/crops: pathogenicity/toxicity, growth inhibition
- non-target species: mammals, other terrestrial vertebrates (for example birds), invertebrates (for example bees, earthworms, springtails), aquatic vertebrates (for example fish), invertebrates (for example benthic, epibenthic) and aquatic plants (for example algae)
Applicants must provide, with appropriate reference(s). Please provide a copy of each publication/report referenced.
9.11 Exposure characterization
Please provide an exposure assessment with the following considerations included:
- given the intended: target crops/plants, intended method of application, rate, timing and frequency of application, conduct direct and indirect exposure assessments for the proposed trial scenario
- direct: worker/bystander by dermal , inhalation and ingestion exposure routes
- indirect: soil, groundwater, plant uptake routes of exposure
9.12 Microbial characterization
Please characterize the microorganisms by providing the following details (please provide references for all information provided):
Natural occurrence
- Geographical distribution
- Natural habitats: soils, water, atmosphere, on or inside of living organisms (for example endophyte, epiphyte)
- Hosts (symbiotic, saprophytic or pathogenic relationships)
Description of the life cycle
- Characteristics of the different forms of the microorganism during its life cycle (for example motile cells, dormant cysts, spores)
- Mechanism for reproduction and dispersal
- Mechanism for survival (in adverse conditions)
Physiological properties
- Growth parameters (for example temperature, pH, osmotic minima, maxima and optima)
9.13 Microorganisms modified by molecular biological techniques
For microorganisms modified by molecular biological techniques, the following information is required:
- flow diagram representing the genetic modification process including:
- the map of the construct inserted in the host
- location (chromosomal or plasmid)
- copy number
- cloning vector(s) used
- promoter sequences
- selectable marker genes including any antibiotic resistance genes
- detailed description of the gene product(s), their properties and functions
- description of the metabolic pathways altered by the insertion
- unintended effects on gene expression (down-regulation or up-regulation of other genes)
- stability of the inserted genetic material
- horizontal gene transfer potential: capacity to transfer the genetic material between the organism and non-target species, the mechanisms of possible transfer (transformation, transduction or conjugation) and the elements involved (plasmids, bacteriophages, integrative conjugative elements, transposons, insertion sequences, integrons, gene cassettes and genomic islands)
- procedures and tests to detect and quantify the modified microorganisms
For more guidance in preparing complete and well-structured safety rationales, please refer to Tab 5 in the Guide to Submitting Applications for Registration under the Fertilizers Act.
9.14 Monitoring plan and procedure
A monitoring plan of the spread and establishment of the novel supplement or fertilizer containing a novel supplement in the environment throughout the duration of the trial must be provided. The methodology used to identify and trace the novel chemical or microorganism must be approved by the CFIA prior to implementation.
Sampling and testing of non-edible and edible plant parts, in cases where the product may lead to residues in/or on the plant/crop (for example in cases of endophytic or epiphytic colonization respectively) may also be required as part of the monitoring plan.
Release of some novel supplements and fertilizers containing novel supplements for scientific research may require that the experimental site be monitored after termination of the trial. In such cases, the sampling protocols and the analytical methods used to detect and monitor the persistence of the product in the environment must be approved by the CFIA prior to trial establishment. Once approved, the monitoring plan and procedures shall be included in the research authorization as 1 of the conditions of the release.
If at any point in time during or after completion of the research project, it is determined that the release of the product has had any adverse effects on plant, animal, and human health or the environment, appropriate measures such as fumigation of the soil and/or post-harvest land restrictions will be implemented at the cost of the applicant and/or responsible researcher.
9.15 Confinement procedures
A detailed description of the confinement procedures intended to mitigate the establishment and spread of the novel supplement or fertilizer containing a novel supplement in the environment must be submitted for research trials conducted both in a greenhouse and in the field. These include but are not limited to descriptions of the packaging of the product during shipment and storage, timing and application methods, implementation of buffer zones (if required), research area entry restrictions, equipment cleanup and sterilization, etc. If deemed necessary the CFIA may impose additional confinement conditions on the trials to mitigate the spread of the novel product.
9.16 Contingency plan
All applicants must provide a contingency plan designed to mitigate any adverse effect(s) of an accidental release of the product. This includes a detailed description of cleanup and disposal procedures in cases of accidental spill or release of the product, treated plant material or growing medium contaminated with the novel supplement or fertilizer containing a novel supplement.
10. Inspection
All research trials on novel supplements and fertilizers containing a novel supplement conducted in Canada may be subject to inspection by CFIA inspectors. The CFIA will generate a detailed inspection report form designed to verify compliance of the trial with all conditions prescribed in the authorization. The CFIA inspectors may require assistance from the researcher/cooperator of the trial to fill out all the required information on the report form. Cooperation of the researchers is greatly appreciated. Following the field inspection, the inspector will send a completed report form to the Fertilizer Safety Section for review and follow up. Any changes to the research protocol must be submitted to and approved by the Fertilizer Safety Section prior to trial inspection.
11. Records and data reporting
In addition to monitoring via data collection, the trials must be monitored by the responsible researcher on a regular basis (minimum frequency of once every 4 weeks). This is to assess the health of the plants, containment of the crop within the field trial plots, and the health of any adjacent vegetation, etc. Treatment plots should also be compared to control plots to identify any potential negative effects of the novel supplement or fertilizer containing a novel supplement. Observations should be recorded accordingly. The CFIA does not provide templates for these observations.
Any unanticipated effect that may be attributed to the release or spread of the novel supplement or the fertilizer containing a novel supplement under test must be reported immediately upon its observation to the CFIA. If any adverse effects associated with the release of the product are noted the CFIA may cancel the research authorization and require corrective actions to mitigate the risks.
12. Contact information
Fertilizer Safety Section
Canadian Food Inspection Agency
Phone: 1-855-212-7695
Email: cfia.paso-bpdpm.acia@inspection.gc.ca
Appendix A: examples of acceptable maps
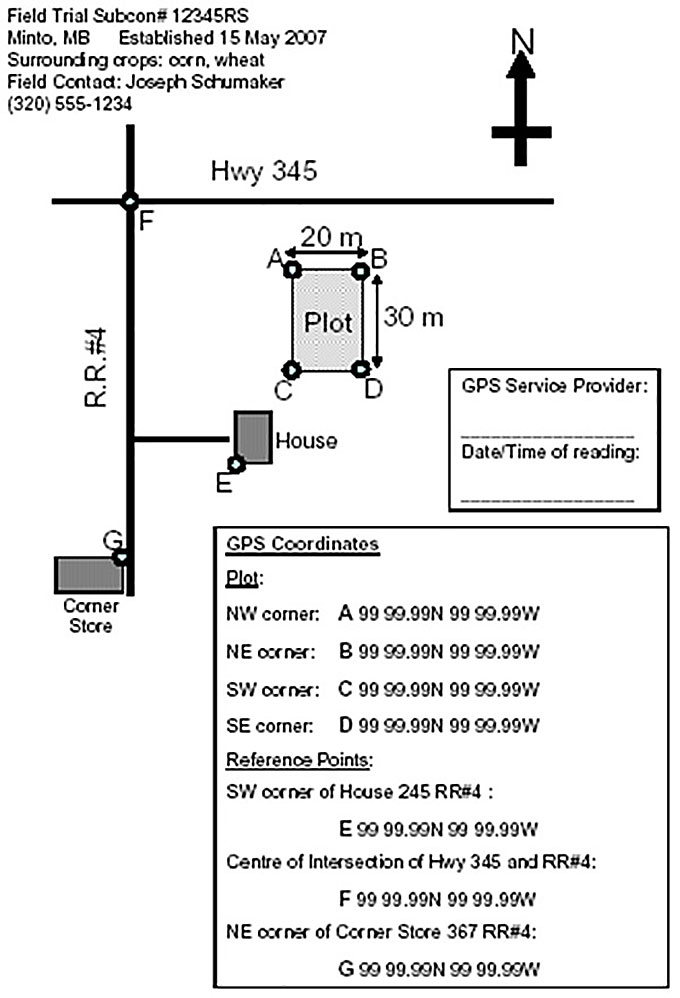
Description of image – Acceptable maps example 1
The following text appears at the top left corner of the image:
Field Trail Subcon# 12345RS
Minto, MB Established 15 May 2007
Surrounding crops: corn, wheat
Field contact: Joseph Schumaker
320-555-1234
Below the text is a map.
At the top there is an arrow pointing up with an N showing that the top of the map is facing north.
The map shows 2 lines that intersect at the top left corner of the map. The vertical line represents R.R.#4 and the horizontal line represents Highway 345.
At the bottom of the R.R.#4 line is a box the represents a corner store and the letter G. Towards the top of the R.R.#4 line where the line intersects Highway 345 is the letter F. Toward the bottom of the R.R.#4 line, there is a short line and at the end of the line there is the letter E that represent a house. Above the house, there is a rectangular that represent the plot which is 20mx30m and a letter (A, B, C, D) in each corner. Next to the house on the right there is a box that contains the following text:
GPS service provider:
Date/Time of reading:
Below the house, there is a box that contains the following text:
GPS coordinates
Plot
NW corner: A 99 99.99N 99 99.99W
NE corner: B 99 99.99N 99 99.99W
SW corner: C 99 99.99N 99 99.99W
SE corner: D 99 99.99N 99 99.99W
Reference points
SW corner of house 245 R.R.#4:
E 99 99.99N 99 99.99W
Centre of intersection of Hwy 345 and R.R.#4:
F 99 99.99N 99 99.99W
NE corner of corner store 367 R.R.#4:
G 99 99.99N 99 99.99W
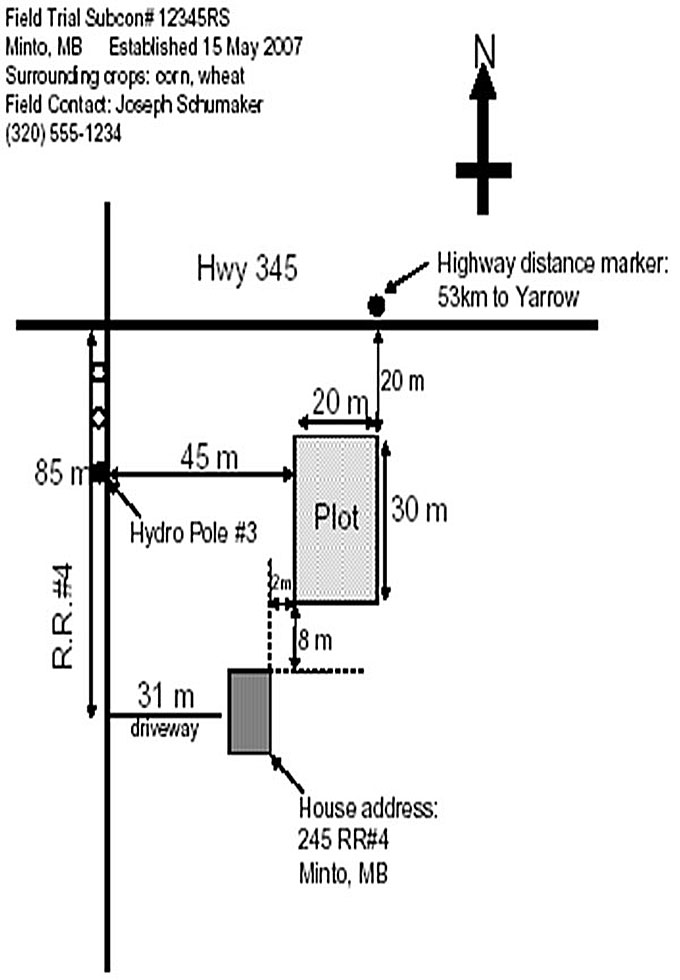
Description of image – Acceptable maps example 2
The following text appears at the top left corner of the image:
Field Trail Subcon# 12345RS
Minto, MB Established 15 May 2007
Surrounding crops: corn, wheat
Field contact: Joseph Schumaker
320-555-1234
Below the text is a map.
At the top there is an arrow pointing up with an N showing that the top of the map is facing north.
The map shows 2 lines that intersect at the top left corner of the map. The vertical line represents R.R.#4 and the horizontal line represents Highway 345.
20 m from Highway 345 on R.R.#4, there is a Hydro Pole #3 and 45m to the right of this pole there is a Plot 20mx30m. 85m from Highway 345 on R.R.#4, there is a driveway of 31m and at the end of the driveway there is house, where the address is 245 R.R.#4, Minto, MB
- Date modified: